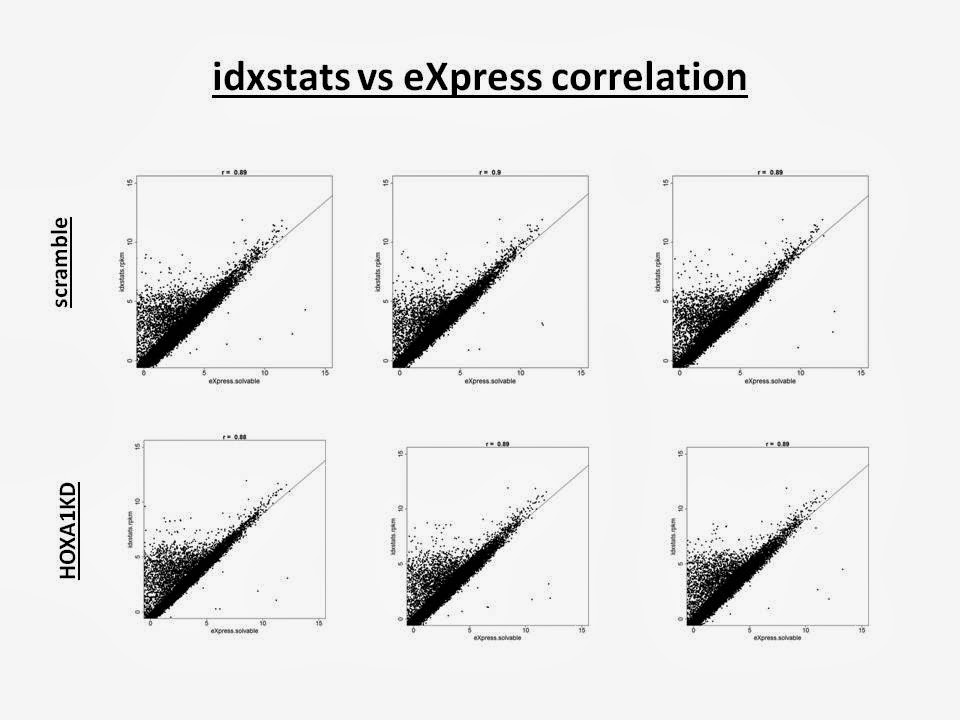
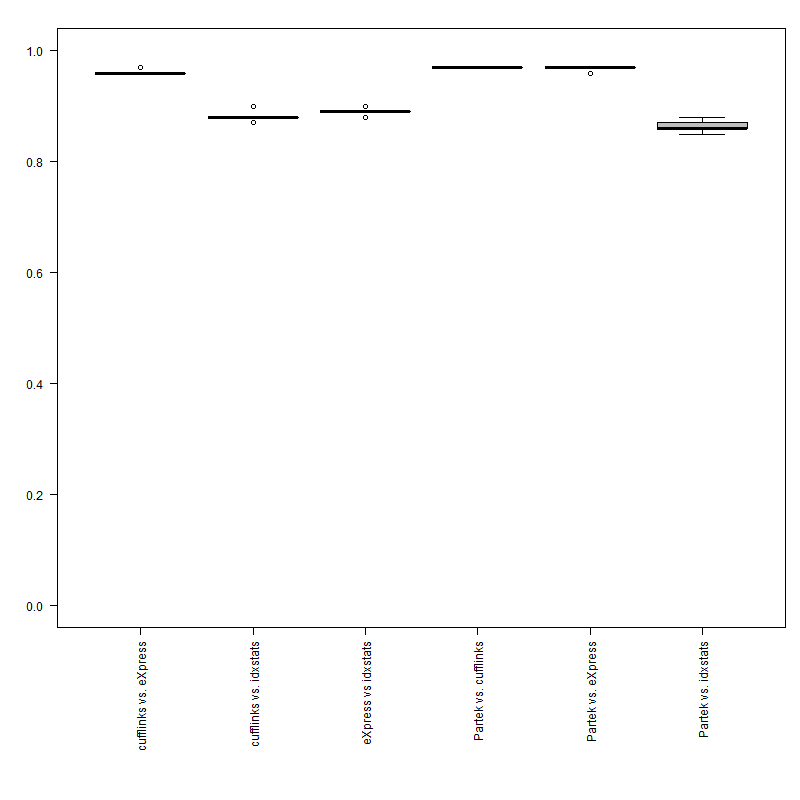Pros:
- Tested lots of programs
- Used several benchmarks
Cons:
- No experimental validation and/or cross-platform/protocol comparison (for example, Figure 6 defines accuracy based upon overlap with known exon junctions).
- I think qPCR validation (or microarray data, spike-ins, etc.) would be useful to compare gene expression levels - for example, see validation from Rapport et al. 2013.
- Limited empirical test data (E-MTAB-1728 for processed values; ERR033015 / ERR033016 for raw data; total n = 2): 1 human cell line sample, 1 mouse brain sample, and simulated data.
- In contrast, I ran differential expression benchmarks (Warden et al. 2013) comparing 2-group comparisons with much more data: patient cohort with over 100 samples (ERP001058) as well as a 2-group cell line comparison with triplicates (SRP012607). Likewise, the cell line results also briefly compared RNA-Seq to microarray data in my paper.
- Accordingly, there are no gene list comparisons, and I think gene expression analysis is probably the most popular type of RNA-Seq analysis
- Used strand-specific protocol - not sure how robust findings are for other protocols. For example, I think a lot of data currently being produced is not strand-specific.
- Only compared at paired end alignments, but (for gene expression analysis) single-end data is probably most common and technically sufficient for gene expression analysis (I can't recall the best possible citation for this, but the Warden et al. paper shows STAR single-end and paired-end to be quite similar). Results may differ for PE versus SE alignments. For example, this was the case with Novoalign but not really the case for STAR; however, to be fair, this particular difference could be determined ahead of time from the Novocraft website.
In practice, I would probably choose between TopHat and STAR (two of the most popular options). I would say that this paper confirms my previous benchmarks showing that these two programs are more or less comparable with each other. When I tested STAR, I noticed some formatting issues: for example, I think the recommended settings weren't sufficient to get it to work with cufflinks, and I think Partek had to do some re-processing to produce the stats in our paper. I assume these problems should be fixable (and I see no technical problem with STAR), but this is why I haven't already switched to using STAR over TopHat on a regular basis.
The result I found potentially interesting is that it seems like STAR may be better than TopHat for variant calling (none of the analysis in the paper that I published can address this question). However, I would want to see some true validation results, and I think that most users are not concerned with this (and even fewer have paired DNA-Seq and RNA-Seq data to distinguish genomic variants from RNA-editing events).
To be fair, I don't think this paper was designed to provide the type of benchmarks I was most interested in seeing. However, I think there was still room to predict testable hypotheses and define accuracy with validation experiments. For example, the authors could have checked how aligners affect splicing events predicted by tools like MATS, MISO, etc. (as long as they produced the samples used in the benchmarks; alternatively, it wouldn't have been too hard to produce some new data for the purpose of being able to perform validation experiments).
Plus, there was a second paper published in the same issue with a number of the same authors (Steijger et al. 2013). So, maybe this paper isn't really meant to be read in isolation. For example, that other paper seems to report considerable discrepancies isoform-level distributions (which matches my own experience that gene-level abundance is preferable for differential expression and splicing event predictions seem more reliable than whole transcript predictions). In short, I would certainly recommend reading both papers - in addition to others like Rapport et al. 2013, Liu et al. 2014, Seyednasrollah et al. 2013, Warden et al. 2013, etc.